Conversation
Co-authored-by: Manuel Blesa <mblesac@gmail.com> Co-authored-by: Paola Galdi <paola.galdi@gmail.com>
Co-authored-by: Manuel Blesa <mblesac@gmail.com> Co-authored-by: Paola Galdi <paola.galdi@gmail.com>
Co-authored-by: Manuel Blesa <mblesac@gmail.com> Co-authored-by: Paola Galdi <paola.galdi@gmail.com>
|
Hi @Lestropie, We've been working on this following your advice. You'll see three commits:
The results are very similar to the original implementation (note that the results are never identical). Let us know what do you think about the changes. In the meantime, I'm doing the paperwork to be able to send you a dataset, then you can do the actual testing. I think your points have been addressed in the different modifications to the code, but there are thee I would like to comment:
I think is still an interesting option to have. The cortical parcellation provided by the dHCP has only 32 nodes. So I don't think it is gonna be used for connectomics. The M-CRIB atlas is the translation of the freesurfer parcellation to the neonatal brain, so I think that will become one of the most used parcellations (with permission of the neonatal AAL derived or the JHU parcellations). My opinion is to leave it as an additional output, the people can always use another parcellation, but in this way, you can save a lot of time.
The initial WM segmentation created is done combining the WM and the sGM of the dHCP outputs (we call it WM, which can be misleading). So in fact we are inserting the sGM of the M-CRIB atlas in the combination of WM+sGM of the dHCP. I found this approach to provide a better delineation of the subcortical structures.
I have no answer for that. These structures are very difficult to segment in this population due to the size/resolution and the low contrast. I think it makes sense to leave it also for these algorithms as it is and let the users decide, but again, I have no definitive answer. |
My question here was not about any potential stochasticity within the relevant ANTs commands (which is what I suspect you're referring to here), but moreso the fact that I observed potential errors in your code relating to duplicate / absent indices, and so hoped that you would spot the differences in script outcomes from those and confirm whether or not their resolution is correct.
While true, it doesn't quite address the original question. Let's say you have a voxel that is outside of the dHCP combined WM & sGM mask; it could be cGM or CSF according to dHCP. Now that voxel is classified as sGM by M-CRIB. What will be the outcome of your script in that voxel?
No worries. I'll pose that question over on the main MRtrix3 repo. |
| DHCP_AMYG_HIPP = list(range(1, 5)) | ||
|
|
||
| MCRIB_CGM = list(range(1000, 1036) + range(2000, 2036)) | ||
| MCRIB_CGM = list(range(1000, 1004)) + list(range(1005,1036)) + list(range(2000, 2004)) + list(range(2005,2036)) |
There was a problem hiding this comment.
This relates to my question RE: absent / duplicated indices. Looks like the elements I thought may have been accidentally omitted you intended to omit; but I have some recollection that there were additionally indices that were duplicated in your original code. This is the better way to do it, but I was partly curious whether or not you would spot the influence of those duplications in a change in output of the script.
(P.S. Given it looks like an unusual omission, it might be worth adding a code comment indicating the structures omitted and why)
There was a problem hiding this comment.
Hi,
We have been working on this, these are the new changes:
- Now the algorithms are called mcrib and dhcp.
- The dHCP algorithm has been modified, now you just need to input the path of the derivates (/anat) folder and that's all, no mask required anymore. Note that there are some versions of the dHCP pipeline around with different names, I think now it covers all the possible cases.
- In the dHCP algorithm we added a filter to ensure that the merging of the sGM doesn't overlap with the CSF.
- A note has been added about the missing labels: the labels 1004 and 2004 don't exist in the original M-CRIB atlas.
As it is now, the dHCP algorithm is the same as in my original paper (with the addition of this CSF filter to ensure that the sGM doesn't enter in the CSF). Basically, it combines the tissue probability maps of the dHCP pipeline with the binary subcortical gray matter parcellations of the M-CRIB atlas. However, I've been thinking to modify it and add two possibilities: the use of the probability maps or not. This will translate in the addition of a new flag (-posteriors), that if specify it it will work as it does now, the only difference will be the use of tissue probability maps for the subcortical GM, in this way all the tissues will have the partial volumes. If not specify it, the algorithm will use only the binary segmentations for everything. I was thinking about this because probably a lot of the people that run the dHCP pipeline didn't use the -additional flag (for example in the dHCP data itself, this data is not there). What do you think? Does this make sense?
…put) Manuel Blesa <mblesac@gmail.com> Paola Galdi <paola.galdi@gmail.com>
|
Diverting conversation from #1 (comment) to main thread as it relates to more items than were highlighted in the code for that specific comment.
If you wanted to be really fancy, you could make parsing of the user-specified input path more robust, so that as long as the user provides a path that can be reasonably interpreted the script will still proceed. So e.g. if the user provides the path to the subject directory, the
The description of the situation is still a bit too ambiguous for me to be able to immediately advise here, as I'm not familiar with the data provided by the pipeline. Let's see if I understand the situation correctly:
If this is the case, then my advice would be that the script should check for presence of tissue probability priors in dHCP data.
However I'm concerned my interpretation is incorrect because you then talk about use of tissue probability priors for sub-cortical GM:
So please correct my dot points above as necessary and I can revise. |
Yes, that's right.
dHCP provides a segmentation of the sGM (also tissue probability priors only if
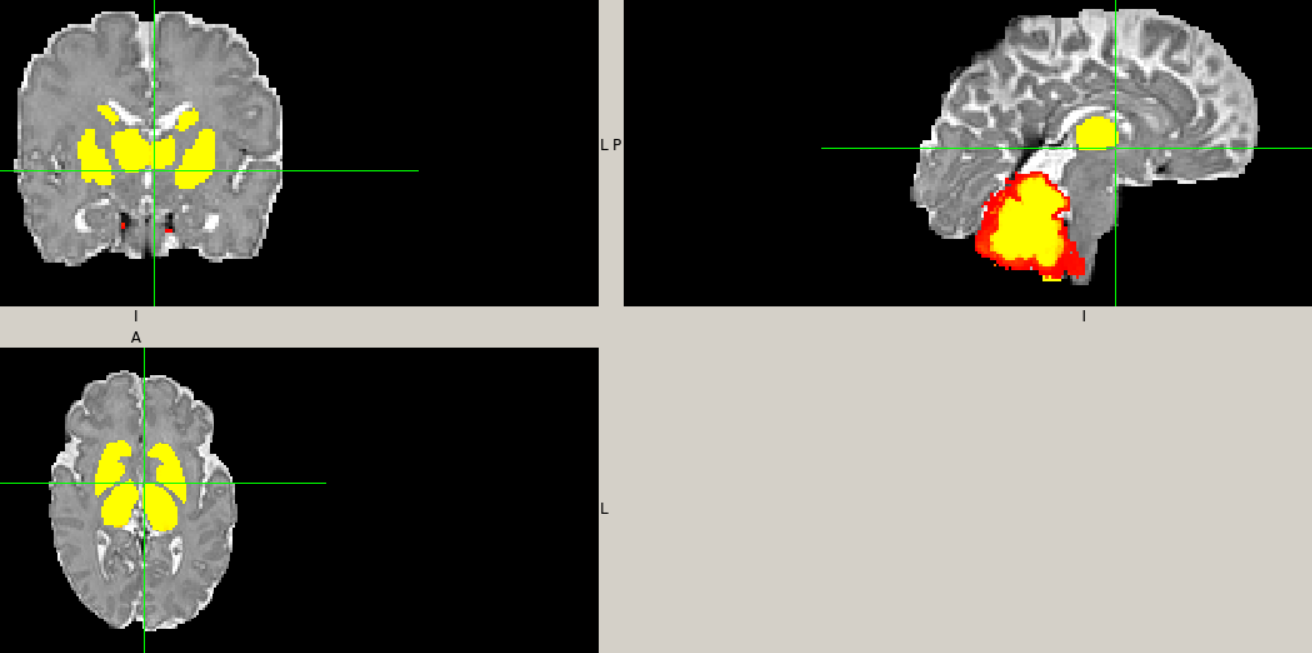
M-CRIB + ANTs provides both, binary and tissue probability priors, but for the original implementation I used the binary masks. Please take a look to an example (ignore the cerebellum): My reason for choosing the M-CRIB subcortical parcellation was that it was not as "bulky" as the dHCP one and it could allow the WM tracts to better pass through the middle. This was purely visual, based on my opinion, I didn't do any quantitative experiment.
What I was thinking is:
Does make sense? |

|
OK cool. So an important detail here is that both dHCP and M-CRIB (may) have non-binary segmentations. I personally don't think there's a huge benefit in offering independent control of the two.
That logic doesn't quite work as it results in issuing a warning if the user specifies that command-line option. It should instead be something like: use_hard_segmentation = False
if app.ARGS.hard:
use_hard_segmentation = True
elif not os.path.isfile(soft_parcellation_path):
app.warn('No tissue posteriors found (-additional flag not used); output will be hard segmentation')
use_hard_segmentation = TrueVariable Note I've changed to "hard segmentation" rather than "binary"; I think that's more faithful to the fact that the output 5TT image is 4D rather than 3D. |
…atal algorithms Co-authored-by: Manuel Blesa <mblesac@gmail.com> Co-authored-by: Paola Galdi <paola.galdi@gmail.com>
|
Hi, I added the option to use the hard or the soft segmentation (by default uses the soft). I tested in a dHCP subject and it looks like this: Now you can test it with any subject of the dHCP and it should work. I'm still waiting for the confirmation from the HR office to be able to send you another subject from our cohort (with all the posteriors) so you can try it there as well. |




- Ensure that input files are unique and unambiguous. - Remove redundant logic across get_inputs() and execute() regarding use of hard segmentation. - Simplify logic of primary conversion loop. - Generate sub-cortical grey matter image from M-CRIB only once, rather than once per tissue; this should facilitate code execution without using the -force option.
|
Also, at some point run the |
Hi @mblesac @paola-g,
I had some fun cleaning up and Pythonifying the code and so now have a bit of a clearer picture as to what's going on. Given I don't yet have any exemplar data I've not made any attempt at all to run it, so there may well be outright syntax errors in there let alone runtime issues; but depending on their severity / how cryptic the errors are you might be able to address some stuff at your end (feel free to commit directly to this branch if you wish). My changes should also clean up the terminal output quite a bit.
This PR proposes merging my new branch on your fork into the
masterbranch of your fork. Therefore, once this PR is merged, the corresponding changes will appear in MRtrix3#2393. So we will use this PR to discuss the cleanup / refactoring between us, and then MRtrix3#2393 can be used for final discussion with a wider audience before merge into the MRtrix3devbranch.I'm tempted to suggest that the dHCP pipeline should be a separate
5ttgenalgorithm.While it's true that the joint label fusion with the M-CRIB data is executed in both scenarios:
I'm unsure of how much utility the output of the
-parcellationoption would be if the dHCP pipeline data are used. Also, if the dHCP version were standalone, it could omit the modality command-line argument, which is non-functional in that use case.Minor points:
Given other work in BIDS & quantitative imaging I prefer to use e.g. "T1w" rather than "T1" wherever possible.
(Though admittedly now that I think of it this is a problem in
5ttgen fsl...)Config file entries are camel case, so I changed "
MCRIBpath" to "MCRIBPath".I'm not sure if the histogram matching is requisite here; I'd have expected the ANTs scripts to be doing their own intensity matching (or using a registration metric that doesn't necessitate such)?
I don't think it is necessarily guaranteed that the sGM segmentations provided by M-CRIB will always lie within voxels that are classified by the dHCP pipeline as containing 100% WM. So greater care might be required regarding how those sGM segmentations are inserted; i.e. it might not be only the WM image that needs to be decreased to accommodate those extra parcels.
I'm strongly considering changing the
-sgm_amyg_hippoption to be specific to the5ttgen fslalgorithm, and to always assign the amygdalae and hippocampi to the sGM volume in other algorithms. The option was put there because in5ttgen fslthe segmentations of those structures by FIRST are on occasion less extensive than the GM fraction estimated by FAST, and so streamlines are not able to reach those structures for the sGM priors to kick in; but it's a problem specific to that algorithm, and I suspect that it should not only be the default behaviour for other algorithms but also that the converse behaviour doesn't even need to be provided. Do you have any thoughts on this based on your own experiences?There's a lot of importing of data into the scratch directory that can probably be omitted. Not only the import as
.mifthen conversion to.nii; much of those data could likely be used in-place without having to convert anything into the scratch directory. You just need to make sure to use absolute filesystem paths (since the user may specify a path relative to the working directory, but the relevant command is executed from inside the scratch directory).Many scratch file names could possibly be shortened in order to reduce the length of terminal outputs.
There were some errors in I think it was the MCRIB GM parcel list: a couple of nodes missing and a couple of nodes duplicated. I hope you can see from the code restructuring how it facilitates diagnosing such things. But it's worth (once it's in an operational state) comparing the output of this version to your own code to confirm.